
Summary
Although most mutations in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome are expected to be deleterious and rapidly cleared or relatively neutral, a small proportion will affect functional properties and may alter infectivity. , the severity of disease, or interactions with the host. immunity. The appearance of SARS-CoV-2 in late 2019 was followed by a period of relative evolutionary stasis that lasted approximately 11 months.
However, since the end of 2020, the evolution of SARS-CoV-2 has been characterized by the appearance of sets of mutations, in the context of “variants of concern”, that affect the characteristics of the virus, including transmissibility and antigenicity, probably in response to the changing immune system profile of the human population. There is emerging evidence of reduced neutralization of some SARS-CoV-2 variants by post-vaccination serum; however, a greater understanding of the correlates of protection is required to assess how this may affect vaccine efficacy.
However, manufacturers are preparing platforms for a possible update of vaccine sequences, and it is critical that monitoring of genetic and antigenic changes in the global virus population be carried out alongside experiments to elucidate the phenotypic impacts of vaccines. mutations. In this review, we summarize the literature on mutations of the spike variant protein of SARS-CoV-2, the primary antigen, focusing on their impacts on antigenicity and contextualizing them in protein structure, and discussing them in the context of frequencies of mutation observed in the world sequence data sets.
SARS-CoV-2 spike variants
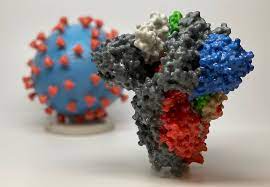
Sites of variation in the SARS-CoV-2 spike protein. Amino acids in bright red have variations in many individuals, pink amino acids vary in fewer individuals, and white amino acids show very few variants. Viruses, in their nonsensical way, are masters of evolution. Two aspects of viral biology make them particularly successful. First, huge populations of viruses are generated as they infect cells and replicate. For example, during the peak of SARS-CoV-2 infection, there may be between 1 and 100 billion viruses in an infected person.
Second, their molecular machinery for replication is often sloppy, introducing occasional errors into the progeny. This is the perfect combination for rapid evolution. During an infection, many variants of the virus can be produced in these populations. Most sequence variations will harm the virus or be neutral with little change for better or worse, but the occasional variant will improve some aspect of the viral life cycle. These rare advantageous variants have emerged several times in SARS-CoV-2 and have caused new waves of infection in the current COVID-19 pandemic.
Variation assessment
Scientists around the world have studied the evolution of SARS-CoV-2 to understand its capabilities and help plan for the future. The illustration shown here maps the main sites of variation of the spike protein, based on more than 3 million samples that have been sequenced and deposited in the GISAID database. The structure is based on PDB ID 7kj2, but the coordinates were taken from SWISS-MODEL since the original PDB entry does not have atomic coordinates for several flex loops. Also, glycosylation is not shown in this illustration, to make protein variation easier to see, so you should imagine the protein covered with multiple carbohydrate chains.
Functional improvements
As you can see, the variation sites are scattered throughout the three-dimensional structure. Scientists are still working out the functions of each of these changes, but some of the more common sites of variation are becoming clearer. The most common mutation (currently at least) is at position 614.
It is believed to control the stability of the top of the spike. Another common mutation, 681, is found in a flexible loop that is clipped by the cellular protease furin, breaking the chain into two pieces. The upper part (S1) recognizes the host cell and the lower part (S2) directs fusion and cell entry. Researchers have found that this cleavage makes the virus more infectious with cells in the respiratory tract.
Variant structures
During the COVID-19 pandemic, SARS-CoV-2 has spread throughout the world and variants have emerged by chance in different countries and spread rapidly from there. Recent variant structures (PDB IDs 7lwv, 7lyo, 7v7q, 7v7e, 7t9k). They all have multiple changes, including sites where an amino acid has mutated (shown in red) and sites where amino acids have been removed from the chain.
They all include the two common changes mentioned above, along with other changes scattered throughout the structure. These can benefit the virus in many ways: mutations in the receptor-binding domain and C-terminal domains can improve recognition and attachment to cells, changes in the N-terminal domain can help evade the immune system and mutations in the S2 region can enhance the process of fusion and cell entry.
Peak variation at position 614
Mutation from aspartate to glycine at position 614 (shown in red) removes an interaction with threonine 859 (turquoise) in a neighbouring subunit in the trimeric peak. This is thought to loosen the structure, facilitating the transition to the active conformation with extended receptor-binding domains. To compare the native structure with aspartate at position 614 (PDB ID 6vyb) and the variant delta-mutated structure with glycine (PDB ID 7v7q).