During the peak of the COVID-19 pandemic in Kazakhstan (June 2020), the media reported multiple cases of SARS-CoV-2 PCR-negative pneumonia with increased mortality. Our objective was to study the epidemiological characteristics of hospitalized patients with positive and negative PCR with analysis of hospital and post-hospital mortality. We also compare the characteristics of respiratory diseases between 2019 and 2020.
Methods
The study population consists of 17,691 (March-July-2020) and 4,600 (March-July-2019) hospitalized patients with respiratory diseases (including COVID-19). Incidence rate, case fatality rate, and survival analysis for overall mortality (in-hospital and post-hospital) were evaluated.
Study population and data sources
The study population consisted of all hospitalized patients with respiratory illnesses (including COVID-19) according to the International Statistical Classification of Diseases and Related Health Problems (ICD-10) from March to July 2019 and from March to July 2020 in Turkestan oblast, Kazakhstan. The following ICD-10 codes were included in the study: J00-J06 (acute upper respiratory tract infections), J09-J18 (influenza and pneumonia), J20-J22 (other acute lower respiratory tract infections), J40- J47 (chronic diseases of the lower respiratory tract), J96-J99 (other diseases of the respiratory system), B34 (viral infection of unspecified site), Z20 (contact and “suspected” exposure to communicable diseases), U07.1 (COVID-19 specified virus) and U07 .2 (COVID-19 unspecified virus).
The raw data was retrieved from the Single National Electronic Health System (UNEHS) linked with the records to the “Electronic Registry of Internal Patients” that included data on dates of admission and discharge, ICD-10 codes, dates and results of PCR tests, discharge results and some demographic data. Global mortality statistics (in-hospital and post-hospital death) were obtained independently from the “Adjunct Population Registry” and were linked to hospitalized patients through the Population Registry Number (RPN-ID); each date of death followed by the date of hospital discharge is considered post-hospital mortality. The population census of the Turkestan oblast, including all cities and rural areas (2,016,100 people), was obtained from the State Statistics Committee.
SARS-CoV-2 infection detection method
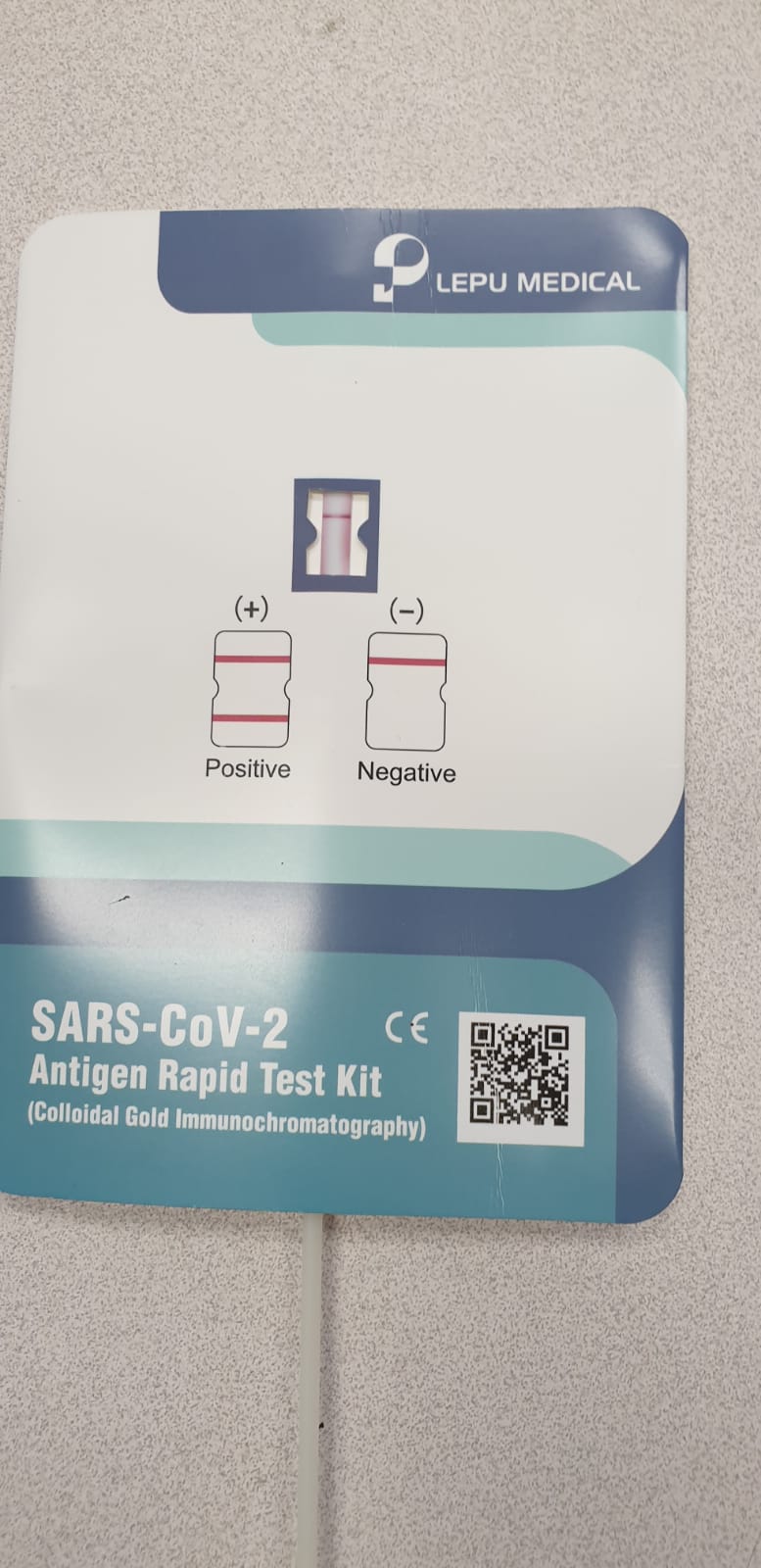
Confirmation of SARS-CoV-2 infection was performed by real-time quantitative PCR on nasopharyngeal swabs with the BGI kit (Beijing Genomics Institute, Shenzhen, China) in defined special regional laboratory settings.
Assessment results
Incidence, mortality and lethality rates were evaluated. Incidence and mortality rates were calculated for each year using the number of newly diagnosed patients and deaths, and the size of the population. The case fatality rate was calculated by dividing the number of deaths by the number of newly diagnosed cases. The incidence was compared by year of admission. All-cause mortality was divided into in-hospital and post-hospital mortality, which was used to identify associated risk factors among admissions in 2020.
The start of follow-up was the date of hospital admission, and patients were followed until death or the end of the follow-up period (August 30, 2020). Two outcome variables were of interest for survival analysis: in-hospital mortality (time from hospital admission to hospital discharge) and overall (in-hospital and post-hospital combined) mortality (time from hospital admission to death at any time up to 30 days). August 2020). ). Censoring for in-hospital mortality survival analysis was taken on the date of hospital discharge, and for pooled mortality, it was August 30, 2020.
Statistic analysis
For each diagnostic group, absolute numbers of hospitalizations and deaths, incidence and mortality rates per 100,000, case fatality rates per year were reported. Absolute and relative frequencies were reported for categorical variables. Means and standard deviations were used to describe continuous variables, while biased continuous variables were characterized by medians and interquartile ranges (IQRs). Parametric bivariate analysis (Pearson’s Chi-squared, two-sample t-test, ANOVA) was used to assess associations of demographic and disease-related characteristics with outcome variables.
Kaplan-Meier survival curves were plotted for the results of the PCR test. Cox proportional hazards models were fitted with epidemiologically and statistically significant covariates using backwards stepwise selection. The proportional hazards assumption for different groups was tested using log plots. We performed a sensitivity analysis to assess the robustness of our main findings.
We examined the association between overall mortality (in-hospital and post-hospital) and sociodemographic parameters in a subgroup of patients admitted to only provisional and infectious disease hospitals (excluding patients who were in quarantine). The significance level of 5% (α < 0.05) was taken. All statistical analyzes were performed using STATA 16.0 statistical software. The study was approved by the Institutional Review Ethics Committee (NU-IREC 203/29112019) with exemption from informed consent.
Results
Respiratory disease incidence and mortality rates were 4 and 11 times higher in 2020 compared to 2019 (877.5 vs. 228.2 and 11.2 vs. 1.2 per 100,000, respectively). PCR-positive cases (compared to PCR-negative) had a two-fold increased risk of overall mortality. We observed a 24% higher risk of death in men than in women and in older patients than in younger ones. Patients residing in rural areas had a 66% higher risk of death compared to city residents, and being treated in a makeshift hospital was associated with 1.9 times higher mortality compared to those treated in hospitals of infectious diseases.
Conclusion
This is the first study from the Central Asia and Eurasia regions, assessing mortality from SARS-CoV-2 PCR positive and PCR negative respiratory system diseases during the peak of the COVID-19 pandemic. We describe a higher mortality rate for PCR-positive cases compared to PCR-negative cases, for men compared to women, for older patients compared to younger patients, and for patients living in rural areas. rural compared to city residents.
Although most mutations in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome are expected to be deleterious and rapidly cleared or relatively neutral, a small proportion will affect functional properties and may alter infectivity. , the severity of disease, or interactions with the host. immunity. The appearance of SARS-CoV-2 in late 2019 was followed by a period of relative evolutionary stasis that lasted approximately 11 months.
However, since the end of 2020, the evolution of SARS-CoV-2 has been characterized by the appearance of sets of mutations, in the context of “variants of concern”, that affect the characteristics of the virus, including transmissibility and antigenicity, probably in response to the changing immune system profile of the human population. There is emerging evidence of reduced neutralization of some SARS-CoV-2 variants by post-vaccination serum; however, a greater understanding of the correlates of protection is required to assess how this may affect vaccine efficacy.
However, manufacturers are preparing platforms for a possible update of vaccine sequences, and it is critical that monitoring of genetic and antigenic changes in the global virus population be carried out alongside experiments to elucidate the phenotypic impacts of vaccines. mutations. In this review, we summarize the literature on mutations of the spike variant protein of SARS-CoV-2, the primary antigen, focusing on their impacts on antigenicity and contextualizing them in protein structure, and discussing them in the context of frequencies of mutation observed in the world sequence data sets.
SARS-CoV-2 spike variants
Sites of variation in the SARS-CoV-2 spike protein. Amino acids in bright red have variations in many individuals, pink amino acids vary in fewer individuals, and white amino acids show very few variants. Viruses, in their nonsensical way, are masters of evolution. Two aspects of viral biology make them particularly successful. First, huge populations of viruses are generated as they infect cells and replicate. For example, during the peak of SARS-CoV-2 infection, there may be between 1 and 100 billion viruses in an infected person.
Second, their molecular machinery for replication is often sloppy, introducing occasional errors into the progeny. This is the perfect combination for rapid evolution. During an infection, many variants of the virus can be produced in these populations. Most sequence variations will harm the virus or be neutral with little change for better or worse, but the occasional variant will improve some aspect of the viral life cycle. These rare advantageous variants have emerged several times in SARS-CoV-2 and have caused new waves of infection in the current COVID-19 pandemic.
Variation assessment
Scientists around the world have studied the evolution of SARS-CoV-2 to understand its capabilities and help plan for the future. The illustration shown here maps the main sites of variation of the spike protein, based on more than 3 million samples that have been sequenced and deposited in the GISAID database. The structure is based on PDB ID 7kj2, but the coordinates were taken from SWISS-MODEL since the original PDB entry does not have atomic coordinates for several flex loops. Also, glycosylation is not shown in this illustration, to make protein variation easier to see, so you should imagine the protein covered with multiple carbohydrate chains.
Functional improvements
As you can see, the variation sites are scattered throughout the three-dimensional structure. Scientists are still working out the functions of each of these changes, but some of the more common sites of variation are becoming clearer. The most common mutation (currently at least) is at position 614.
It is believed to control the stability of the top of the spike. Another common mutation, 681, is found in a flexible loop that is clipped by the cellular protease furin, breaking the chain into two pieces. The upper part (S1) recognizes the host cell and the lower part (S2) directs fusion and cell entry. Researchers have found that this cleavage makes the virus more infectious with cells in the respiratory tract.
Variant structures
During the COVID-19 pandemic, SARS-CoV-2 has spread throughout the world and variants have emerged by chance in different countries and spread rapidly from there. Recent variant structures (PDB IDs 7lwv, 7lyo, 7v7q, 7v7e, 7t9k). They all have multiple changes, including sites where an amino acid has mutated (shown in red) and sites where amino acids have been removed from the chain.
They all include the two common changes mentioned above, along with other changes scattered throughout the structure. These can benefit the virus in many ways: mutations in the receptor-binding domain and C-terminal domains can improve recognition and attachment to cells, changes in the N-terminal domain can help evade the immune system and mutations in the S2 region can enhance the process of fusion and cell entry.
Peak variation at position 614
Mutation from aspartate to glycine at position 614 (shown in red) removes an interaction with threonine 859 (turquoise) in a neighbouring subunit in the trimeric peak. This is thought to loosen the structure, facilitating the transition to the active conformation with extended receptor-binding domains. To compare the native structure with aspartate at position 614 (PDB ID 6vyb) and the variant delta-mutated structure with glycine (PDB ID 7v7q).
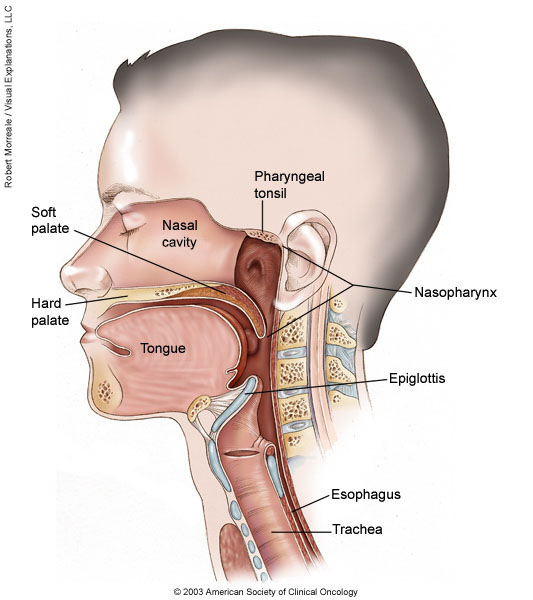
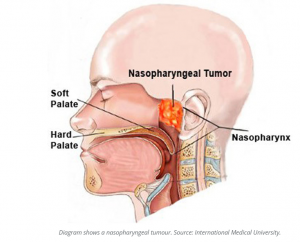
Nasopharyngeal carcinoma is a cancer that occurs in the nasopharynx, which is located behind the nose and above the back of the throat. Nasopharyngeal carcinoma is rare in the United States. It occurs much more frequently in other parts of the world, specifically in Southeast Asia.
Nasopharyngeal carcinoma is difficult to detect early. This is probably because the nasopharynx is not easy to examine and the symptoms of nasopharyngeal carcinoma are similar to other more common conditions. Treatment for nasopharyngeal carcinoma usually involves radiation therapy, chemotherapy, or a combination of the two. You can work with your doctor to determine the exact approach for your particular situation.
Symptoms
In its early stages, nasopharyngeal carcinoma may not cause any symptoms. Possible notable symptoms of nasopharyngeal carcinoma include:
A lump in the neck caused by a swollen lymph node
blood in your saliva
Bloody discharge from the nose
Nasal congestion or ringing in the ears
Hearing loss
Frequent ear infections
Throat pain
Headaches
When to see a doctor
Early symptoms of nasopharyngeal carcinoma may not always prompt you to see your doctor. However, if you notice unusual and persistent changes in your body that don’t seem right to you, such as unusual nasal congestion, see your doctor.
Causes
Cancer begins when one or more gene mutations cause normal cells to grow out of control, invade surrounding structures, and eventually spread (metastasize) to other parts of the body. In nasopharyngeal carcinomas, this process begins in the squamous cells that line the surface of the nasopharynx.
It is not known exactly what causes the genetic mutations that lead to nasopharyngeal carcinoma, although factors, such as the Epstein-Barr virus, have been identified that increase the risk of this cancer. However, it is not clear why some people with all risk factors never develop cancer, while others with no apparent risk factors do.
Risk factor’s
Researchers have identified some factors that seem to increase the risk of developing nasopharyngeal carcinoma, including:
Sex. Nasopharyngeal carcinoma is more common in men than in women.
Race. This type of cancer most commonly affects people in parts of China, Southeast Asia, and North Africa. In the United States, Asian immigrants have a higher risk of this type of cancer than Asians born in the United States. Alaskan Eskimos are also at increased risk of nasopharyngeal cancer.
Years. Nasopharyngeal cancer can occur at any age, but it is most often diagnosed in adults between the ages of 30 and 50.
Salt-cured foods. Chemicals released in the steam when cooking salt-cured foods, such as canned fish and vegetables, can enter the nasal cavity, increasing the risk of nasopharyngeal carcinoma. Being exposed to these chemicals at a young age can further increase the risk.
Epstein Barr virus. This common virus usually produces mild signs and symptoms, like those of a cold. It can sometimes cause infectious mononucleosis. The Epstein-Barr virus is also linked to several rare cancers, including nasopharyngeal carcinoma.
Family history. Having a relative with nasopharyngeal carcinoma increases the risk of developing the disease.
Alcohol and tobacco. Excessive alcohol consumption and tobacco use can increase the risk of developing nasopharyngeal carcinoma.
Complications
Complications of nasopharyngeal carcinoma can include:
Cancer that grows to invade nearby structures. Advanced nasopharyngeal carcinoma can cause complications if it grows large enough to invade nearby structures, such as the throat, bones, and brain.
Cancer has spread to other areas of the body. Nasopharyngeal carcinoma often spreads (metastasizes) beyond the nasopharynx.
Most people with nasopharyngeal carcinoma have regional metastases. This means that cancer cells from the original tumour have migrated to nearby areas, such as the lymph nodes in the neck. Cancer cells that spread to other areas of the body (distant metastases) most often travel to the bones, lungs, and liver.
Prevention
There is no sure way to prevent nasopharyngeal carcinoma. However, if you are concerned about your risk of nasopharyngeal carcinoma, you may want to consider avoiding habits that have been associated with the disease. For example, you can choose to reduce the amount of salt-cured foods you eat or avoid these foods altogether.
Tests to detect nasopharyngeal carcinoma
In the United States and other areas where the disease is rare, routine screening for nasopharyngeal carcinoma is not done. But in areas of the world where nasopharyngeal carcinoma is much more common—for example, in some areas of China—doctors may offer screening to people thought to be at high risk for the disease. Screening may include blood tests for the Epstein-Barr virus.
Primary angle-closure glaucoma (PACG) is estimated to have an effect on over 30 million individuals worldwide by 2040 and is extremely prevalent in the Asian inhabitants. PACG is extra extreme and carries 3 times the upper threat of blindness than major open-angle glaucoma, thus representing a big public well being concern. High heritability and ethnic-specific predisposition to PACG recommend the involvement of genetic elements in illness improvement. In the latest previous, genetic research have led to the profitable identification of a number of genes and loci related to PACG throughout totally different ethnicities.
The exact mobile and molecular roles of those a number of loci in the event and development of PACG stays to be elucidated. Nonetheless, these research have considerably elevated our understanding of the rising mobile processes and organic pathways that may present extra vital insights into the illness’s genetic etiology and could also be helpful for future scientific purposes. This assessment goals to summarize and replace the present information of PACG genetics evaluation analysis.
Identification of Protein Direct Interactome with Genetic Code Expansion and Search Engine OpenUaa
Protein crosslinks happen endogenously equivalent to modifications by ubiquitin-like proteins for signaling, or exogenously by way of genetically encoded chemical crosslinkers (GECX) for finding out elusive protein-protein interactions. However, it stays difficult to determine these protein crosslinks effectively on the proteomic scale. Herein, software program OpenUaa is developed for figuring out protein crosslinks generated by genetically encoded unnatural amino acids and endogenous protein conjugation.
OpenUaa options inclusive and open search functionality, dramatically bettering identification sensitivity and protection. Integrating GECX with OpenUaa, the direct interactome of thioredoxin is recognized in Escherichia coli cells, yielding 289 crosslinked peptides and similar to 205 direct binding protein of thioredoxin. These recognized direct binders present proof for thioredoxin’s regulation of redox state and mitochondria power metabolism.
When figuring out endogenous conjugation of small ubiquitin-like modifier (SUMO), OpenUaa additionally markedly improves protection of SUMOylated peptides by ≈92%, revealing new SUMO targets. GECX-OpenUaa will allow environment friendly identification of direct interactomes of assorted proteins in stay cells.
Progress on animal domestication beneath inhabitants genetics
Animal domestication is the method of adjusting wild animals into domesticated animals that may be stored stably for an extended time period. As the content material of the Neolithic agricultural revolution, domestication is among the vital milestones of the progress of human civilization. Due to the shut relationship between people and animals, domestication has not solely modified the wild state of animals, but in addition modified the habits and historic processes of human beings.
The key query on animal domestication analysis embrace who’s the ancestors of the domesticated animals have been, the modifications produced by domestication, and the time and place of domestication. Due to the advances in high-throughput genomic applied sciences and correspondence evaluation strategies, animal domestication is usually studied on the inhabitants stage.
Here we talk about the analysis content material of animal domestication beneath inhabitants genetics, together with inhabitants historical past, choice indicators, in addition to gene introgression, and we spotlight two new broaden contents, particularly, courting the preliminary time of gene choice and the time of gene introgression. Finally, we summarize the latest analysis progress of main domesticated together with pig, hen, sheep and goat. These advances present a brand new insights and perspective for the analysis on the animal domestication.
Sparse deep neural networks on imaging genetics for schizophrenia case-control classification
Deep studying strategies maintain robust promise for figuring out biomarkers for scientific software. However, present approaches for psychiatric classification or prediction don’t enable direct interpretation of unique options. In the current examine, we introduce a sparse deep neural community (DNN) method to determine sparse and interpretable options for schizophrenia (SZ) case-control classification.
An L0 -norm regularization is applied on the enter layer of the community for sparse characteristic choice, which might later be interpreted primarily based on significance weights. We utilized the proposed method on a big multi-study cohort with grey matter quantity (GMV) and single nucleotide polymorphism (SNP) knowledge for SZ classification.
A complete of 634 people served as coaching samples, and the classification mannequin was evaluated for generalizability on three unbiased datasets of various scanning protocols (N = 394, 255, and 160, respectively). We examined the classification energy of pure GMV options, in addition to mixed GMV and SNP options. Empirical experiments demonstrated that sparse DNN barely outperformed unbiased element evaluation + help vector machine (ICA + SVM) framework, and extra successfully fused GMV and SNP options for SZ discrimination, with a median error fee of 28.98% on exterior knowledge.
The significance weights instructed that the DNN mannequin prioritized to pick out frontal and superior temporal gyrus for SZ classification with excessive sparsity, with parietal areas additional included with decrease sparsity, echoing earlier literature. The outcomes validate the applying of the proposed method to SZ classification, and promise prolonged utility on different knowledge modalities and traits which finally might outcome in clinically helpful instruments.
Comprehensive genetic evaluation of adhesin proteins and their position in virulence of Candida albicans
Candida albicans is a microbial fungus that exists as a commensal member of the human microbiome and an opportunistic pathogen. Cell surface-associated adhesin proteins play a vital position in C. albicans’ skill to bear mobile morphogenesis, develop strong biofilms, colonize, and trigger an infection in a number. However, a complete evaluation of the position and relationships between these adhesins has not been explored. We beforehand established a CRISPR-based platform for environment friendly era of single- and double-gene deletions in C. albicans, which was used to assemble a library of 144 mutants, comprising 12 distinctive adhesin genes deleted singly, and each attainable mixture of double deletions.
Here, we exploit this adhesin mutant library to discover the position of adhesin proteins in C. albicans virulence. We carry out a complete, high-throughput display screen of this library, utilizing Caenorhabditis elegans as a simplified mannequin host system, which recognized mutants crucial for virulence and vital genetic interactions. We carry out follow-up evaluation to evaluate the flexibility of high- and low-virulence strains to bear mobile morphogenesis and kind biofilms in vitro, in addition to to colonize the C. elegans host.
We additional carry out genetic interplay evaluation to determine novel vital detrimental genetic interactions between adhesin mutants, whereby combinatorial perturbation of those genes considerably impairs virulence, greater than anticipated primarily based on virulence of the only mutant constituent strains. Together, this examine yields vital new perception into the position of adhesins, singly and in combos, in mediating various sides of virulence of this crucial fungal pathogen.
Sex chromosome aneuploidies (SCAs) happen in 1 in each 400 births. SCAs are extremely variable and have unsure prognoses, complicating the supply of prenatal cell-free DNA (cfDNA) outcomes or analysis following amniocentesis or chorionic villus sampling. Using a mixed-methods strategy, we explored the experiences of mother and father receiving a prenatal analysis of a fetus with SCA. Responses to open-ended questions had been qualitatively analyzed. Of the 323 mother and father who accomplished the survey, 122 obtained a prenatal analysis and answered no less than one open-ended query.
Most mother and father didn’t recall being knowledgeable that cfDNA screening or amniocentesis might reveal the presence of a SCA previous to testing and described feeling unprepared for a optimistic end result. Variation was discovered between mother and father who had been delivered a analysis by a genetic skilled versus different scientific specialties. Many mother and father expressed that the analysis was delivered in a means that emphasised the unfavorable attributes of the SCA and that they had been supplied restricted help supplies.
Parents who obtained a prenatal analysis of a SCA expressed a need for extra supportive supply of prenatal analysis that focuses on parental training and nuanced dialogue of potential phenotypes. Genetic counselors needs to be conscious of the vary of parental experiences when receiving a SCA analysis from non-genetic suppliers. Prenatal SCA diagnoses are predicted to extend as prenatal cfDNA screening turns into extra broadly used. Collaborations for higher supplier training and complete supplies on SCAs are important to facilitate the supply of SCA diagnoses and enhance guardian understanding and help.
Inference of inhabitants genetic parameters from an irregular time collection of seasonal influenza virus sequences
Basic abstract statistics that quantify the inhabitants genetic construction of influenza virus are essential for understanding and inferring the evolutionary and epidemiological processes. However, the sampling dates of international virus sequences within the final a number of a long time are scattered nonuniformly all through the calendar. Such temporal construction of samples and the small efficient measurement of viral inhabitants hampers the use of typical strategies to calculate abstract statistics.
Here, we outline statistics that overcome this downside by correcting for the sampling-time distinction in quantifying a pairwise sequence distinction. A easy linear regression technique collectively estimates the mutation price and the extent of sequence polymorphism, thus offering an estimate of the efficient inhabitants measurement. It additionally results in the definition of Wright’s FST for arbitrary time-series information. Furthermore, as a substitute for Tajima’s D statistic or the site-frequency spectrum, a mismatch distribution corrected for sampling-time variations could be obtained and in contrast between precise and simulated information.
Application of these strategies to seasonal influenza A/H3N2 viruses sampled between 1980 and 2017 and sequences simulated underneath the mannequin of recurrent optimistic choice with metapopulation dynamics allowed us to estimate the synonymous mutation price and discover parameter values for choice and demographic construction that match the remark. We discovered that the mutation charges of HA and PB1 segments earlier than 2007 had been notably excessive and that together with recurrent optimistic choice in our mannequin was important for the genealogical construction of the HA section. Methods developed right here could be typically utilized to inhabitants genetic inferences utilizing serially sampled genetic information.
Atypical Genetic Basis of Pyrazinamide Resistance in Mono-resistant Mycobacterium tuberculosis
Pyrazinamide (PZA) is a broadly used antitubercular chemotherapeutic. Typically, PZA resistance (PZA-R) emerges in M. tuberculosis strains with current resistance to isoniazid and rifampicin (MDR) and is conferred by loss-of-function pncA mutations that inhibit conversion to its energetic kind, Pyrazinoic acid (POA). PZA-R departing from this canonical situation is poorly understood. Here, we genotype pncA and purported various PZA-R genes (panD, rpsA, and clpC1) with long-read sequencing of nineteen phenotypically PZA mono-resistant isolates collected in Sweden and evaluate their phylogenetic and genomic traits to a giant set of MDR PZA-R (MDRPZA-R) isolates. We report the primary affiliation of ClpC1 mutations with PZA-R in scientific isolates, within the ClpC1 promoter (clpC1p-138) and N-terminal (ClpC1Val63Ala).
Mutations have emerged in each these areas underneath POA choice in vitro and ClpC1N-terminal has been implicated additional, by its POA-dependent efficacy in PanD proteolysis. ClpC1Val63Ala mutants spanned 4 Indo-oceanic sublineages. Indo-oceanic isolates invariably harbored ClpC1Val63Ala and had been starkly overrepresented (OR=22.2, p <0.00001) amongst PZA mono-resistant isolates (11/19) in comparison with MDRPZA-R isolates (5/80). The genetic foundation of Indo-oceanic isolates’ overrepresentation in PZA mono-resistant TB stays undetermined, however substantial circumstantial proof suggests ClpC1Val63Ala confers low-level PZA resistance. Our findings spotlight ClpC1 as doubtlessly clinically related for PZA-R and reinforce the significance of genetic background within the trajectory of resistance growth.
Exploring mother and father’ perceptions of the worth of pediatric genetic counseling affected person letters: A qualitative research presenting classes realized
Genetic counseling affected person letters are a helpful complement to genetic counseling follow. As the demand for genetic companies will increase, enhancing effectivity in each day duties resembling letter writing might enhance genetic counselor workflow. Additionally, understanding the worth recipients place on the content material of these letters previous to creating efficiencies is important towards making certain that the utility of these letters will not be misplaced. To higher perceive mother and father’ perceptions of the letter’s worth within the pediatric genetic counseling setting, we employed a qualitative design involving 13 mother and father of youngsters who obtained a affected person letter following their analysis.
Parents participated in a semi-structured focus group, interview, or cellphone interview, and the info had been analyzed utilizing thematic evaluation. In addition to gathering perceptions of their kid’s letter, we sought to study preferences for letter size, formatting, and stage of element by asking for verbal and written suggestions on three totally different letter codecs created for a fictional affected person. We used self-determination concept (SDT) framework to create the pattern letters, which states that a person’s expertise of autonomy, competence, and relatedness can influence their capacity to interact in actions.
This consists of caring for a baby with particular medical wants. While the findings from this work strengthened the significance of written communication for sufferers as seen in earlier analysis, this work uncovered three main themes in regards to the letter’s worth: (a) parts resembling readability and content material influence guardian emotions of autonomy and enhance competence shifting ahead with their kid’s care; (b) mother and father worth written acknowledgment of the emotional influence of the analysis; and (c) mother and father use the letter as a instrument to speak their kid’s analysis with others. These outcomes can be utilized for creating understandable affected person letters that help autonomy, competence, and relatedness.
Breast fibroepithelial lesions are biphasic tumors which comprise the frequent benign fibroadenomas (FAs) and the rarer phyllodes tumors (PTs). This examine analyzed 262 (42%) standard FAs, 45 (7%) mobile FAs, and 321 (51%) benign PTs contributed by the International Fibroepithelial Consortium, utilizing a beforehand curated 16 gene panel. Benign PTs have been discovered to own a better variety of mutations, and larger charges of most cancers driver gene alterations than each teams of FAs, specifically MED12, TERT promoter, RARA, FLNA, SETD2, RB1, and EGFR.
Cases with MED12 mutations have been additionally extra more likely to have TERT promoter, RARA, SETD2, and EGFR. There have been no vital differences detected between standard FAs and mobile FAs, apart from PIK3CA and MAP3K1. TERT promoter alterations have been most optimum in discriminating between FAs and benign PTs. Our examine affirms the function of sequencing and key mutations that will help in refining diagnoses of those lesions.
From allozymes to NGS: inhabitants genetics of forest bushes in Slovakia prior to now 40 years
This evaluation summarizes the event of inhabitants genetics and inhabitants genomics research of forest bushes in Slovakia in the course of the previous 40 years. Various protein and DNA markers have been utilized throughout this era to deal with a number of matters in evolutionary genetics and biogeography of bushes: allozymes, uniparentally inherited chloroplast and mitochondrial markers, easy sequence repeats and single nucleotide polymorphisms.
The principal object of research of phylogeny and postglacial migration have been Fagus sylvatica s.l. and eastern-Mediterranean firs (Abies Mill. part Abies), the place the divergence of genetic lineages (species and subspecific taxa) in time, in addition to colonization of the present ranges in the course of the Holocene have been reconstructed. The research on intraspecific gene movement and homoploid hybridization centered on hybrid swarms Pinus sylvestris/P. mugo and firs. Unusual maternal inheritance of chloroplast DNA was revealed in P. mugo × P. sylvestris crosses.
Contrasting geographical constructions of hybrid zones have been revealed in wind-dispersed vs. animal-dispersed bushes. Within the research of adaptation, alerts of choice have been recognized each in discipline observations and common-garden experiments on Picea abies, F. sylvatica and A. alba. Perspectives of ongoing analysis using next-generation sequencing have been shortly outlined.
Structural elements of rod opsin and their implication in genetic illnesses
Vision in dim-light situations is triggered by photoactivation of rhodopsin, the visible pigment of rod photoreceptor cells. Rhodopsin is manufactured from a protein, the G protein coupled receptor (GPCR) opsin, and the chromophore 11-cis-retinal. Vertebrate rod opsin is the GPCR finest characterised on the atomic stage of element.
Since the discharge of the primary crystal construction 20 years in the past, an enormous variety of constructions have been launched that, together with worthwhile spectroscopic determinations, unveiled most elements of the photobleaching course of. Numerous spontaneous mutations of rod opsin have been discovered linked to vision-impairing illnesses like autosomal dominant or autosomal recessive retinitis pigmentosa (adRP or arRP, respectively) and autosomal congenital stationary evening blindness (adCSNB). While adCSNB is especially attributable to constitutive activation of rod opsin, RP reveals extra variegate determinants affecting completely different elements of rod opsin operate.
The overwhelming majority of missense rod opsin mutations impacts folding and trafficking and is linked to adRP, an incurable illness that awaits gentle on its molecular construction determinants. This evaluation article summarizes all main structural info out there on vertebrate rod opsin conformational states and the insights gained up to now into the structural determinants of adCSNB and adRP linked to rod opsin mutations. Strategies to design small chaperones with therapeutic potential for chosen adRP rod opsin mutants will probably be mentioned as effectively.
Action detection utilizing a neural community elucidates the genetics of mouse grooming habits
Automated detection of complicated animal behaviors stays a difficult drawback in neuroscience, notably for behaviors that include disparate sequential motions. Grooming is a prototypical stereotyped habits and is commonly used as an endophenotype in psychiatric genetics. Here, we used mouse grooming habits for instance and developed a common function neural community structure able to dynamic motion detection at human observer-level efficiency and working throughout dozens of mouse strains with excessive visible variety.
We present insights into the quantity of human annotated coaching information which are wanted to attain such efficiency. We surveyed grooming habits within the open discipline in 2,457 mice throughout 62 strains, decided its heritable elements, carried out GWAS to stipulate its genetic structure, and carried out PheWAS to hyperlink human psychiatric traits through shared underlying genetics. Our common machine studying resolution that robotically classifies complicated behaviors in giant datasets will facilitate systematic research of behavioral mechanisms.
Shared genetic structure throughout psychiatric problems
Psychiatric problems overlap considerably on the genetic stage, with family-based strategies lengthy pointing towards transdiagnostic threat pathways. Psychiatric genomics has progressed quickly within the final decade, shedding gentle on the organic make-up of cross-disorder threat at a number of ranges of research. Over 100 genetic variants have been recognized that have an effect on a number of problems, with many extra to be uncovered as pattern sizes proceed to develop.
Cross-disorder mechanistic research construct on these findings to cluster transdiagnostic variants into significant classes, together with in what tissues or when in improvement these variants are expressed. At the upper-most stage, strategies have been developed to estimate the general shared genetic sign throughout pairs of traits (i.e. single-nucleotide polymorphism-based genetic correlations) and subsequently mannequin these relationships to determine overarching, genomic threat components.
Description: The Bioperfectus Monkeypox Virus Real Time PCR Kit is an in vitro diagnostic test, based on real-time PCR technology, for the detection of DNA from the Monkeypox virus. Specimens can be obtained from human serum, lesion exudate samples and scab. BSL-2 facilities with standard BSL-2 work practices may be used for the test of t he Monkeypox virus.
Description: Monkeypox virus is the virus that causes the disease monkeypox in both humans and animals. Monkeypox virus is an Orthopoxvirus, a genus of the family Poxviridae that contains other viral species that target mammals. The virus is mainly found in tropical rainforest regions of central and West Africa. The primary route of infection is thought to be contact with the infected animals or their bodily fluids. The genome is not segmented and contains a single molecule of linear double-stranded DNA, 185000 nucleotides long. The Monkeypox Virus real time PCR Kit contains a specific ready-to-use system for the detection of the Monkeypox Virusthrough polymerase chain reaction (PCR) in the real-time PCR system. The master contains reagents and enzymes for the specific amplification of the Monkeypox Virus DNA. Fluorescence is emitted and measured by the real time systems ́ optical unit during the PCR. The detection of amplified Monkeypox Virus DNA fragment is performed in fluorimeter channel 530nm with the fluorescent quencher BHQ1. DNA extraction buffer is available in the kit and serum or lesion exudate samples are used for the extraction of the DNA. In addition, the kit contains a system to identify possible PCR inhibition by measuring the 560nm fluorescence of the internal control (IC). An external positive control defined as 1×10^7 copies/ml is supplied which allow the determination of the gene load.
Description: Monkeypox virus is the virus that causes the disease monkeypox in both humans and animals. Monkeypox virus is an Orthopoxvirus, a genus of the family Poxviridae that contains other viral species that target mammals. The virus is mainly found in tropical rainforest regions of central and West Africa. The primary route of infection is thought to be contact with the infected animals or their bodily fluids.The genome is not segmented and contains a single molecule of linear double-stranded DNA, 185000 nucleotides long.The Monkeypox Virus real time PCR Kit contains a specific ready-to-use system for the detection of the Monkeypox Virusthrough polymerase chain reaction (PCR) in the real-time PCR system. The master contains reagents and enzymes for the specific amplification of theMonkeypox VirusDNA. Fluorescence is emitted and measured by the real time systems ́ optical unit during the PCR. The detection of amplified Monkeypox Virus DNA fragment is performed in fluorimeter channelFAM with the fluorescent quencher BHQ1. DNA extraction buffer is available in the kit and serum or lesion exudate samples are used for the extraction of the DNA. In addition, the kit contains a system to identify possible PCR inhibition by measuring the HEX/VIC/JOE fluorescence of the internal control (IC). An external positive control defined as 1×107copies/ml is supplied which allow the determination of the gene load.
Description: Creative Biogene Monkeypox Virus Real Time PCR Kit is used for the detection of monkeypox Virus in serum or lesion exudate samples by using real time PCR systems. Monkeypox virus (MPV) is a double-stranded DNA, zoonotic virus and a species of the genus Orthopoxvirus in the family Poxviridae. It is one of the human orthopoxviruses that includes variola (VARV), cowpox (CPX), and vaccinia (VACV) viruses. The kit contains a specific ready-to-use system for the detection of the monkeypox Virus. Fluorescence is emitted and measured by the real time systems' optical unit during the PCR.
These components can subsequently be related to exterior traits (e.g. practical imaging phenotypes) to start to know the make-up of those transdiagnostic threat components. As psychiatric genomic efforts proceed to develop, we will start to realize even higher perception by together with extra fine-grained phenotypes (i.e. symptom-level information) and explicitly contemplating the atmosphere. The end result of those efforts will assist to tell bottom-up revisions of our present nosology.
Nonalcoholic fatty liver illness (NAFLD) is characterised by hepatic lipid accumulation. SAMM50 encodes Sam50, a mitochondrial outer membrane protein concerned in the elimination of reactive oxygen species, mitochondrial morphology, and regulation of mitophagy. Certain single nucleotide polymorphisms (SNPs) of SAMM50 have been reported to be correlated with NAFLD.
However, the contribution of SAMM50 polymorphisms to the prevalence and severity of fatty liver in the Chinese Han cohort has not often been reported. Here, we investigated the affiliation between SAMM50 polymorphisms (rs738491 and rs2073082) and NAFLD in a Chinese Han cohort, in addition to the mechanistic foundation of this affiliation. Clinical data and blood samples had been collected from 380 NAFLD instances and 380 regular topics for the detection of genotypes and biochemical parameters. Carriers of the rs738491 T-allele or rs2073082 G-allele of SAMM50 exhibit elevated susceptibility to NAFLD (OR=1.39; 95% CI=1.14-1.71, P=0.001; OR=1.31; 95% CI=1.05-1.62, P=0.016, respectively) and are correlated with elevated serum TG, ALT, and AST ranges.
The presence of the T allele (TT+CT) of rs738491 (P<0.01) or G allele (AG+GG) of rs2073082 (P=0.03) is correlated with the severity of fatty liver in the NAFLD cohort. In vitro research indicated that SAMM50 gene polymorphisms lower its expression and SAMM50 deficiency outcomes in elevated lipid accumulation due to a lower in fatty acid oxidation. Overexpression of SAMM50 enhances fatty acid oxidation and mitigates intracellular lipid accumulation. Our outcomes affirm the affiliation between the SAMM50 rs738491 and rs2073082 polymorphisms and the danger of fatty liver in a Chinese cohort. The underlying mechanism could also be associated to decreased fatty acid oxidation attributable to SAMM50 deficiency.
Cross-species transcriptomics uncovers genes underlying genetic lodging of developmental plasticity in spadefoot toads
That hardcoded genomes can manifest as plastic phenotypes responding to environmental perturbations is a captivating function of residing organisms. How such developmental plasticity is regulated on the molecular stage is starting to be uncovered aided by the event of -omic strategies. Here, we evaluate the transcriptome-wide responses of two species of spadefoot toads with differing capability for developmental acceleration of their larvae in the face of a shared environmental danger: pond drying.
By evaluating gene expression profiles over time and performing cross-species community analyses, we recognized orthologues and purposeful gene pathways whose environmental sensitivity in expression have diverged between species. Genes associated to lipid, ldl cholesterol and steroid biosynthesis and metabolism make up most of a module of genes environmentally responsive in one species, however canalized in the opposite. The evolutionary adjustments in the regulation of the genes recognized by means of these analyses might have been key in the genetic lodging of developmental plasticity in this technique.
Development of Host-Orthogonal Genetic Systems for Synthetic Biology
The building of a host-orthogonal genetic system can’t solely decrease the affect of host-specific nuances on fine-tuning of gene expression, but in addition broaden mobile features equivalent to in vivo steady evolution of genes based mostly on an error-prone DNA polymerase. It represents an rising highly effective strategy for making biology simpler to engineer.
In this evaluation, the latest advances are described on the design of genetic methods that may be stably inherited in the host cells and are answerable for necessary organic processes together with DNA replication, RNA transcription, protein translation, and gene regulation. Their functions in artificial biology are summarized and the longer term challenges and alternatives are mentioned in growing such methods.
Investigating the inhabitants construction and genetic variety of Arabian horses in Oman utilizing SNP markers
Arabian horses had been chosen for metabolic effectivity, magnificence, effectivity and endurance. Therefore, Bedouins have for hundreds of years traced their prized horses’ ancestries. With the institution of the World Arabian Horse Organization (WAHO), registration of Arabian horses grew to become centralized and international locations worldwide registered them in its database.
Most current Arabian horses in Oman right this moment had been imported after the 1970s and are predominantly flat-racing Arabians. This work geared toward revealing the genetic background and variety of Omani Arabian horses by evaluating them with Arabian horses from a various genetic background. To that finish, we genotyped 63 randomly sampled Arabian horses from Oman utilizing the Illumina Equine SNP70. For comparability, SNP genotypes of 12 Saudi Arabian horses, 27 French, 77 Egyptian, 11 Polish and 36 US Arabians had been included in the examine. We moreover included 17 Thoroughbred horses and 21 horses representing giant and small breeds as an outgroup. Our MDS evaluation and phylogenetic evaluation confirmed that the Arabian horses in Oman cluster primarily with French Arabian horses, with a number of horses clustering inside the Polish/US Arabians.
The French Arabian horse cluster was the closest to the Thoroughbred horses. Amongst the Arabian horses, plink common genomic inbreeding ranges had been highest in the Egyptian Arabian (0.169) adopted by the Saudi Arabian horses (0.137) and lowest in the Omani and French Arabian horses, -0.041 and -0.079 respectively. To our data, that is the primary report on the genetic background and variety of Arabian horses in Oman. Our outcomes demonstrated a particular subpopulation construction amongst Arabian horses and this data ought to advise future decision-making on Arabian horse breeding.
A generalized sturdy allele-based genetic affiliation check
The allele-based affiliation check, evaluating allele frequency distinction between case and management teams, is regionally strongest. However, utility of the classical allelic check is restricted in apply, as a result of the tactic is delicate to the Hardy-Weinberg equilibrium (HWE) assumption, not relevant to steady traits, and never straightforward to account for covariate impact or pattern correlation.
To develop a generalized sturdy allelic check, we suggest a brand new allele-based regression mannequin with particular person allele because the response variable. We present that the rating check statistic derived from this sturdy and unifying regression framework incorporates a correction issue that explicitly adjusts for potential departure from HWE, and encompasses the classical allelic check as a particular case.
When the trait of curiosity is steady, the corresponding allelic check evaluates a weighted distinction between individual-level allele frequency estimate and pattern estimate the place the load is proportional to a person’s trait worth, and the check stays legitimate beneath Y-dependent sampling. Finally, the proposed allele-based technique can analyze a number of (steady or binary) phenotypes concurrently and multi-allelic genetic markers, whereas accounting for covariate impact, pattern correlation and inhabitants heterogeneity. To assist our analytical findings, we offer empirical proof from each simulation and utility research. This article is protected by copyright. All rights reserved.
Type I interferons (IFNs) are so-named as a result of they intrude with viral an infection in vertebrate cells. The research of mobile responses to type I IFNs led to the discovery of the JAK-STAT signaling pathway, which additionally governs the response to different cytokine households. We overview right here the end result of viral infections in mice and humans with engineered and inborn deficiencies, respectively, of (i) IFNAR1 or IFNAR2, selectively disrupting responses to type I IFNs, (ii) STAT1, STAT2, and IRF9, additionally impairing mobile responses to type II (for STAT1) and/or III (for STAT1, STAT2, IRF9) IFNs, and (iii) JAK1 and TYK2, additionally impairing mobile responses to cytokines aside from IFNs.
An image is rising of higher redundancy of human type I IFNs for protecting immunity to viruses in pure situations than was initially anticipated. Mouse type I IFNs are important for defense towards a broad vary of viruses in experimental situations. These findings counsel that numerous type I IFN-independent mechanisms of human cell-intrinsic immunity to viruses have but to be found.
Genetic mechanisms and correlational choice construction trait variation in a coral snake mimic
Covariation amongst traits shapes each phenotypic evolution and ecological interactions throughout area and time. However, rampant geographical variation in the energy and route of such correlations could be notably tough to clarify via generalized mechanisms. By integrating inhabitants genomics, surveys of pure historical past collections and spatially express analyses, we examined a number of drivers of trait correlations in a coral snake mimic that reveals exceptional polymorphism in mimetic and non-mimetic color traits.
We discovered that though such traits co-occur extensively throughout area, correlations have been greatest defined by a combination of genetic structure and correlational choice, quite than by any single mechanism. Our findings counsel that spatially complicated trait distributions could also be pushed extra by the easy interplay between a number of processes than by complicated variation in one mechanism alone. These interactions are notably vital in mimicry techniques, which continuously generate hanging geographical variation and genetic correlations amongst color sample traits.
Evaluating the function of GSTP1 genetic polymorphism (rs1695, 313A>G) as a predictor in cyclophosphamide-induced toxicities
The affiliation between Glutathione S-transferase Pi 1(GSTP1) genetic polymorphism (rs1695, 313A>G) and cyclophosphamide-induced toxicities has been broadly investigated in earlier research, nonetheless, the outcomes have been inconsistent. This research was carried out to additional elucidate the affiliation.A complete search was performed in PubMed, Embase, Web of Science, China National Knowledge Infrastructure, and Wan Fang database as much as January 5, 2020. Risk ratios (RRs) and 95% confidence intervals (95% CIs) have been used to estimate the affiliation between GSTP1 rs1695 polymorphism and cyclophosphamide-induced hemotoxicity, gastrointestinal toxicity, an infection, and neurotoxicity.A complete of 13 research have been finally included.
Compared with the GSTP1 rs1695 AA genotype carriers, sufferers with AG and GG genotypes had an elevated threat of cyclophosphamide-induced gastrointestinal toxicity (RR, 1.61; 95% CI, 1.18-2.19; P = .003) and an infection (RR, 1.57; 95% CI, 1.00-2.48; P = .05) in the total inhabitants. In the subgroup analyses, there have been vital associations between GSTP1 rs1695 polymorphism and the threat of cyclophosphamide-induced myelosuppression (RR, 2.10; 95% CI, 1.60-2.76; P < .00001), gastrointestinal toxicity (RR, 1.77; 95%CI, 1.25-2.53; P = .001), and an infection (RR, 2.01; 95% CI, 1.14-3.54; P = .02) in systemic lupus erythematosus (SLE) or lupus nephritis syndrome sufferers, however not in most cancers sufferers.
Our outcomes confirmed a necessary function for the GSTP1 rs1695 polymorphism in the prediction of cyclophosphamide-induced myelosuppression, gastrointestinal toxicity, and an infection in SLE or lupus nephritis syndrome sufferers. More research are essential to validate our findings in the future.
Chromosomal and symbiosis-related genotypes of rhizopine-producing and non-producing isolates of Rhizobium meliloti and Rhizobium leguminosarum have been examined by multilocus enzyme electrophoresis and RFLP. The distribution of rhizopine manufacturing in each species was discovered to be unbiased of host genotype. Conversely, rhizopine manufacturing was related with specific symbiotic plasmid varieties.
This affiliation could clarify the noticed distribution of rhizopine manufacturing in R. leguminosarum and R. meliloti. Rhizopine synthesis (mos) genes confirmed higher sequence divergence than rhizopine catabolism (moc) genes in each R. meliloti and R. leguminosarum. Furthermore, mos and moc genes have been much less divergent in R. leguminosarum than R. meliloti, suggesting a newer evolution in the former species.
The Polymorphism Analyses of Short Tandem Repeats as a Basis for Understanding the Genetic Characteristics of the Guanzhong Han Population
The brief tandem repeat (STR) loci are polymorphic markers in the mixed DNA index system (CODIS) and non-CODIS STR loci. Due to the extremely polymorphic attribute of STR loci, they’re fashionable and broadly used in forensic DNA typing laboratories. In this research, 22 STR loci (1 CODIS, 21 non-CODIS STR loci) and an Amelogenin locus have been genotyped and analyzed in 590 unrelated people of the Guanzhong Han inhabitants. None of the 22 STR loci deviated from the Hardy-Weinberg equilibrium, and all the loci have been in the linkage equilibrium state.
We noticed 247 alleles, and the corresponding allelic frequencies ranged from 0.0008 to 0.3695 in the Guanzhong Han inhabitants. The mixed energy of discrimination and the cumulative exclusion likelihood was 0.999 999 999 999 999 999 999 999 999 346 36 and 0.999 999 999 709 74, respectively. The outcomes together with Nei’s DA genetic distance, multidimensional scaling evaluation, and principal part evaluation confirmed that the Guanzhong Han inhabitants has nearer genetic affinities with Northern Han, Chengdu Han, and Xinjiang Hui teams from China primarily based on allelic frequencies of 15 overlapped STR loci from Guanzhong Han and 13 reference teams.
Description: Clone your gene of interest into this AAV Expression Vector, then co-transfect along with AAV packaging vectors into a packaging host cell line such as 293AAV.
Description: Pre-made over-expression lentivirus for human target: CTAG1B, containing a RFP-Blasticidin dual selection marker.
×
The current outcomes indicated that Microreader™ 23sp ID equipment included extremely polymorphic loci, and it might be effectively used for particular person identification, paternity testing, and inhabitants genetics in the Guanzhong Han inhabitants.
Recently, a number of genome-wide affiliation research recognized PHACTR1 as key locus for 5 numerous vascular issues: coronary artery disease, migraine, fibromuscular dysplasia, cervical artery dissection and hypertension. Although these signify important threat components or comorbidities for ischemic stroke, PHACTR1 function in mind small vessel ischemic disease and ischemic stroke most vital survival mechanism, such because the recruitment of mind collateral arteries like posterior speaking arteries (PcomAs), stays unknown.
Therefore, we utilized exome and genome sequencing in a multi-ethnic cohort of 180 early-onset unbiased familial and apparently sporadic mind small vessel ischemic disease and CADASIL-like Caucasian patients from US, Portugal, Finland, Serbia and Turkey and in 2 C57BL/6J stroke mouse fashions (bilateral widespread carotid artery stenosis [BCCAS] and center cerebral artery occlusion [MCAO]), characterised by totally different levels of PcomAs patency. We report three very uncommon coding variants in the small vessel ischemic disease-CADASIL-like cohort (p.Glu198Gln, p.Arg204Gly, p.Val251Leu) and a stop-gain mutation (p.Gln273*) in one MCAO mouse.
These coding variants do not cluster in PHACTR1 identified pathogenic domains and are not prone to play a critical function in small vessel ischemic disease or mind collateral circulation. We additionally exclude the chance that replicate quantity variants (CNVs) or a variant enrichment in Phactr1 could also be related to PcomA recruitment in BCCAS mice or linked to numerous vascular traits (cerebral blood move pre-surgery, PcomA measurement, leptomeningeal microcollateral size and junction density throughout mind hypoperfusion) in C57BL/6J mice, respectively.
Genetic variability in PHACTR1 is not prone to be a standard susceptibility issue influencing small vessel ischemic disease in patients and PcomA recruitment in C57BL/6J mice. Nonetheless, uncommon variants in PHACTR1 RPEL domains could affect the stroke final result and are price investigating in a bigger cohort of small vessel ischemic disease patients, totally different ischemic stroke subtypes and with purposeful research.
Insight of fetal to grownup hemoglobin change: Genetic modulators and therapeutic targets
The medical heterogeneity of β-hemoglobinopathies is so variable that it prompted the researchers to determine the genetic modulators of those ailments. Though the first modulator is the kind of β-globin mutation which impacts the diploma of β-globin chain synthesis, the co-inheritance of α-thalassemia and the fetal hemoglobin (HbF) ranges additionally act as potent secondary genetic modifiers.
As elevated HbF ranges ameliorate the severity of hemoglobinopathies, in this evaluate, the genetic modulators mendacity inside and outdoors the β-globin gene cluster with their believable function in governing the HbF ranges have been summarised, which in future could act as potential therapeutic targets.
Genetic affiliation of MMP14 promoter variants and their purposeful significance in gallbladder most cancers pathogenesis
Gallbladder most cancers (GBC) is comparatively uncommon however exhibits excessive frequency in sure geographical areas and ethnic teams, which embrace Northern and Eastern states of India. Previous research in India have indicated the doable function of genetic predisposition in GBC pathogenesis. Although matrix metalloproteinase-14 (MMP14) is identified modulator of tumour microenvironment and tumorigenesis and TCGA information additionally suggests its upregulation but, its function in genetic predisposition for GBC is utterly unknown.
We explored MMP14 promoter genetic variants as threat components and their implication in expression modulation and the pathogenesis of GBC. We genotyped all single nucleotide polymorphisms of MMP14 promoter by Sanger’s sequencing in roughly 300 GBC and 300 management examine topics of Indian ethnicity and, in 26 GBC tissue samples. Protein expression of MMP14 in GBC tissue samples was checked by immunohistochemistry. In vitro luciferase reporter assay was carried out to elucidate function of promoter genetic variants on expression ranges in two totally different cell traces.
MMP14 promoter variants, rs1003349 (p worth = 0.0008) and rs1004030 (p worth = 0.0001) have been considerably related to GBC. Luciferase reporter assay confirmed excessive expression for threat alleles of each the SNPs. Genotype-phenotype correlation for rs1003349 and rs1004030, in affected person pattern, confirmed that threat allele carriers had increased expression ranges of MMP14; furthermore, the correlation sample matched with genetic affiliation fashions. Overall, this examine unravels the affiliation of MMP14 promoter SNPs with GBC which contribute to pathogenesis by rising its expression.
Compact Genetic Algorithm-based Feature Selection for Sequence-based Prediction of Dengue-Human Protein Interactions
Dengue Virus (DENV) an infection is one of many quickly spreading mosquito-borne viral infections in people. Every yr, round 50 million folks get affected by DENV an infection, ensuing in 20,000 deaths. Despite the latest experiments specializing in dengue an infection to know its performance in the human physique, a number of functionally vital DENV-human protein-protein interactions (PPIs) have remained unrecognized. This article presents a mannequin for predicting new DENV-human PPIs by combining totally different sequence-based options of human and dengue proteins just like the amino acid composition, dipeptide composition, conjoint triad, pseudo amino acid composition, and pairwise sequence similarity between dengue and human proteins.
A Learning vector quantization (LVQ)-based Compact Genetic Algorithm (CGA) mannequin is proposed for characteristic subset choice. CGA is a probabilistic approach that simulates the habits of a Genetic Algorithm (GA) with lesser reminiscence and time necessities. Prediction of DENV-human PPIs is carried out by the weighted Random Forest approach because it is discovered to carry out higher than different classifiers. We have predicted 1013 PPIs between 335 human proteins and 10 dengue proteins.
Description: Leukocyte or tumor cell interactions with vascular endothelium consist of a cascade of processes including the firm attachment of cells to endothelial cell adhesion molecules. The CytoSelect Tumor Endothelium Adhesion Assay provides a robust system for the quantitative determination of interactions between tumor cells and endothelium. Adherent cells can be easily quantified on a fluorescence plate reader.
Description: Our CytoSelect 384-Well Cell Transformation Assay uses a modified soft agar 3D matrix to support the formation of colonies by neoplastic cells. Quantitation of cell transformation is performed on a fluorescence plate reader.
Description: Our CytoSelect 384-Well Cell Transformation Assay uses a modified soft agar 3D matrix to support the formation of colonies by neoplastic cells. Quantitation of cell transformation is performed on a fluorescence plate reader.
Description: Cell Biolabs? CytoSelect Cell Proliferation Assay Reagent (Fluorometric) provides a fluorometric format for measuring and monitoring cell proliferation. Cells can be plated and then treated with compounds or agents that affect proliferation. Cells are then incubated with the proliferation reagent. Upon entering metabolically active live cells, the non-fluorescent proliferation reagent is converted into a bright red fluorescent form. An increase in cell proliferation is accompanied by increased fluorescent signal, while a decrease in cell proliferation (and signal) can indicate the toxic effects of compounds or suboptimal culture conditions. The assay principles are basic and can be applied to most eukaryotic cell lines, including adherent and non-adherent cells and certain tissues. This cell proliferation reagent can be used to detect proliferation in bacteria, yeast, fungi, protozoa as well as cultured mammalian and piscine cells. The kit contains sufficient reagents for the evaluation of 960 assays in ten 96-well plates or 192 assays in eight 24-well plates.
Description: The ability of malignant tumor cells to invade normal surrounding tissue contributes in large part to the morbidity and mortality of cancers. Cell invasion requires several distinct cellular functions including adhesion, motility, detachment, and extracellular matrix proteolysis. Our CytoSelect Cell Invasion Assays utilize precoated inserts to assay the invasive properties of tumor cells. Invasive cells can be quantified in 24-well plates on either a standard microplate reader or a fluorescence plate reader. Inserts are precoated on the top of the membrane with Laminin.
All predicted interactions are validated by literature filtering, GO-based evaluation, and KEGG Pathway enrichment evaluation. This examine will encourage the identification of potential targets for simpler anti-dengue drug discovery.
What is a comparison of EST databases from other species and tissues?
This shows the diversity in programming sequences between plants along with also a worldwide view about the similarities in enzymes for certain cells or conditions. Nonetheless, the true number of genes present in Arabidopsis, rice, or some other sequenced species remains to be established via functional genomic experiments which establish the biological significance of DNA sequences, because gene forecast through homology comparisons and applications tools is a statistical”best informed guess” instead of a biologically based procedure. For genetic analysis and molecular reproduction of crops, we have to extract DNA in the target plants initially, then execute PCR reactions. High-throughput sequencing technologies has led sequencing of roughly 800 chloroplast genomes from other plants 3 2 conserved areas from plastid (chloroplast) genome (matk+rbcl) were suggested as barcode primers to discriminate large set of angiosperms. The world has seen a rapid gain in the understanding of the plant genome sequences as well as the molecular and physiological purpose of plant enzymes, which has revolutionized the genetics and its efficacy. MinION sequencing is superior to conventional procedures of PCR identification, provided its creation of entire genome sequences that permit the identification of this plant virus strain if it becomes divergent, since it’s not biased with primers that rely upon virus strings. Ribosomal sequences are a goal for analyzing inter- and – intra-species phylogenetics for three years 9 The important design for genotyping-by-sequencing with ribosomal sequences was designing primers about the conserved regions of the ribosomal strings (26S, 5.8S, and 18S) that interval the conserved internal transcribed spacers (ITS). To begin with, most genes are functionally redundant, as even species using easy genomes like Arabidopsis carry extensive duplications, and instant, mutations in several genes might be highly pleiotropic, which could mask the part of a receptor in a particular pathway (Springer, 2000). Yet osmosis is regarded as a part of the toolbox, and it has an significant role in assigning functions.
To examine this theory, we genotyped SAIL_232 along with two randomly chosen lines of the identical collection (SAIL_59 and SAIL_107) with primers specific to the benchmark Col-0 CS70000 genome along with the SAIL-inverted condition (see Methods). PCR analysis revealed that this inversion” was common to each of 3 separate SAIL-lines examined and absent in Col-0 CS70000.
Hence the event wasn’t on account of this T-DNA mutagenesis, rather is a good illustration of the genetic drift”happening during the propagation of the Columbia benchmark” accession within individual labs 30. Production of comprehensive DNA database (utilizing next generation sequencing) while focusing more conserved regions are effective for medicinal plants identification 18,19 These documents would likewise be of help to examine the taxonomy, ecology, phylogeny and morphology of unique species 20 But, the growth of new protocols and amplification approaches with fresh primer cocktails would greatly simplifies the subject of DNA barcoding by constituting more thorough genome data from various species. A good illustration of a bigger, comparatively less intricate genome meeting is the harvest species Brassica rapa 64 An estimated 72× sequencing coverage of the genome was created, equivalent to Illumina shotgun paired-end information from NGS libraries with insert sizes ranging from 200 bp to 10 kb, also constructed with SOAPdenovo 63 The resultant assembly was created from 14,207 contigs larger than 2 kb, further constructed into 794 scaffolds, totalling approximately 283.8 Mb and anticipated to cover over 98 percent of the receptor distance, according to alignments of 214,425 B. rapa people EST sequences and 52,712 unigenes in the BrGP database 65 Further evaluation of the ethics of this assembly was conducted by aligning BAC clone Sanger sequences reported in prior research.
These datasets provide information for creating tools to detect genes for programs in diagnostics and breeding. Companies like Illumina (which recently bought Pacific Biosciences) and 10X Genomics are supplying technology to permit PCR gear to generate long notes of hereditary sequences that offer a more complete image of a genome. Six responses using every one of six AD primers and a boundary primer are utilized to maximize the probability of creating a product. SNP discovery incontestably created a quantum leap ahead with the dawn of NGS technology and massive numbers of SNPs are now accessible from several genomes such as big and intricate types (see Section 4). Unlike model systems like Arabidopsis and people, SNPs from harvest plants remain restricted for now, but accessibility to price NGS promises to boost SNP detection in addition to the creation of reference genome sequences. SNPs are implemented in areas as varied as individual forensics two and diagnostics 3, aquaculture 4, mark assisted-breeding of milk cattle 5, harvest development 6, conservation 7, and source management in fisheries 8 Functional genomic research have bestowed upon SNPs found within regulatory genes, transcripts, and Expressed Sequence Tags (ESTs) 9, 10 Until lately large scale SNP detection in crops was confined to maize, Arabidopsis, and rice 11 – 15 Genetic applications like linkage mapping, and population structure, institution research, map-based cloning, marker-assisted plant breeding, and functional genomics continue to be allowed by access to large collections of SNPs.
The genomes of both parents were sequenced to 10× policy for every single (~5-Gb Illumina re-sequencing information ), while both pools were sequenced to ~20× policy for every single (~10-Gb information; Table 1). Utilizing the genome sequences of Chiifu” since the reference, the reads were both aligned and SNP and insertion/deletion (InDel) variations from the genomes of both parents were predicted. The study of large genomic sequences from other plant species revealed that the frequency of SSRs in Arabidopsis (each 6-7 kb) holds for different plants too. SSR supply in crops: Of 52 DNA sequences over 10 kb in length from species other than Arabidopsis, 38 have been found to possess a minumum of one SSR motif. Extended DNA sequencing reads (up to 2 Mb) enable improved genome meeting with whole characterisation of complex genomic areas — such as structural variations, transposons, and transgene insertions — providing fresh insights into plant biology, development, and breeding approaches. In crops, SNPs are beneficial in species source, connection and scientific studies, the cloning of target loci breeding of genes linkage disequilibrium analysis, DNA fingerprinting, and the building of high resolution maps. The RoI and the HGAP contigs of those three PacBio libraries have been merged separately to an S. verrucosum VER54 chromosome 10 scaffold comprising two called R-gene coding areas to find out if longer fit dimensions can catch the area between R-genes in which promoters and terminator sequences could live.
From time to time, the presence of metabolites in medicinal plants influence DNA caliber during isolation as well as closely related species might demand different DNA isolation protocols 14 The arrangement variation in reference sequence and phylogenetic reconstruction is the simple principle for species identification from crops 15 The use of DNA based markers (except RFLP) as universal primers have important benefits in species identification because they result in great amplification across distinct genomic regions among divergent species 16 Next production sequencing is just another centre of innovative genomics era to have a more exact image of species genome and to identify greater orthologous and paralogous regions at several loci of unique species. Molecular techniques also have been used to examine genetic diversity and evolutionary roots in populations of several different fungal genera (two ). Mitochondrial rRNA genes grow quickly and may be helpful in the ordinal or household level (41). The evolutionary lineage of this oomycetes was elucidated by sequencing studies using small-subunit rRNA sequences (9). Thus far, 951 GWASs are reported in people (? Those technologies have been characterized by the concurrent sequencing of atoms of DNA (instead of clusters”), hence avoiding phasing problems, and the subsequent sequences have a tendency to be from the kb range, providing the chance to build genomes and creating more contigs by surrounding complicated and conserved genomic regions and permitting comparatively high-confidence assemblies of reads.
The launch of draft mention genomes have generally contained major landmarks and have been shown to be invaluable for the research and characterization of genome structure, genes and their expression, diversity and development 1 – 5 The growth of sequence data in a increasing number of taxa has led to comparative research as well as the execution of molecular cloning and biotechnology methods for crop development , 7 The building of the initial plant genomes was made possible by using considerable funds, coordination and attempt to allowing automated Sanger-based sequencing engineering and computational calculations. Rice genes very similar to famous disease resistant genes revealed no cross-hybridization with corn genomic DNA, implying sequence divergence or their lack in maize (Tarchini et al., 2000). There are reports of linearity throughout the mono-dicotyledoneous branch between Arabidopsis and cereals that diverged up to as 200 million decades ago (Mayer et al., 2001) Exploiting colinearity will help establish cross-species genetic connections and also aids from the extrapolation of data from species with easier genomes (i.e. rice) to complex species (wheat, corn ). Advances in high-throughput sequencing have altered genetics and genomics, together with lesser prices resulting in an explosion in genome sequencing project size 1 and amount of species two Genomes from several diverse organisms are sequenced, from marsupials to microbes, plants, phytoplankton, and parasites, one of others 3 For a little while it’s been possible for one lab to string and de novo construct a intricate genome.
While these observations are confined to the transformation vectors, we especially looked in the individual junctions involving genome and also T-DNA to test for epigenetic influences on the flanking genomic DNA sequences/genes. Evaluation of expression and epigenetic signatures about the corresponding T-DNA arrangement is recorded from genome browser shots such as SALK_059379 plasmid pROK2 (c) along with SAIL_232 plasmid pCSA110 (Id ): Illumina read mapping of bisulfite sequencing, RNA-seq and distinct small RNA species. Plant genome technology employing the soil microorganism Agrobacterium tumefaciens has altered plant agriculture and science by allowing testing and identification of chemical functions and providing a mechanism to equip plants with exceptional traits 1, 2, 3 Transport DNA (T-DNA) insertional mutant projects are run in significant dicot and monocot versions, and more than 700,000 lines with receptor affecting insertions are made in Arabidopsis thaliana (Arabidopsis henceforth) independently (examined in’Malley 4). Targeted T-DNA sequencing procedures were conducted approximately 325,000 of those lines to recognize the tumultuous transgene insertions and also to connect genotype with phenotype 4 That abundance of sequence information, a lot of that was made available before publication, is accessible at:, continues to be iteratively updated since 2001, also obtained from the neighborhood around 10 million times by 2018. Arabidopsis thaliana was the first plant genome sequenced 16 followed shortly after by rice 17, 18 At the year 2011 alone, the amount of plant genomes sequenced climbed compared to the amount sequenced in the prior decade, leading to now, 31 and counting, publicly published sequenced plant genomes (). Together with the ever growing throughput of next-generation sequencing (NGS), de novo and reference-based SNP detection and program are now possible for many plant species.
Why Next-generation DNA sequencing has substantially improved our comprehension of the total structure and dynamics of several plant genomes?
The acute limitations still stay because next-generation DNA sequencing reads normally are shorter compared to Sanger reads. This type of approach was successfully studied in barley BAC clones chosen according to BAC-unigene associations explained in that exact same study, thus indicating that BAC swimming sequencing may be utilised in correlation with present physical maps to match or proper whole-genome sequencing assemblies, offering in the process the chance of greater quality contig sequence assemblies in gene-rich areas of plant genomes. De novo assembly of genomes has closely mimicked the tendencies and advancements in sequencing technology and accompanying sequencing assembly applications over recent years 45 The development of next-generation sequencing technology has enabled a far bigger quantity of plant genomes to be sequenced and constructed than that which could have been deemed potential with Sanger sequencing independently, largely due to the costs and labour involved with these endeavors. Though a number of these areas correspond to tandem repeats like telomeric sequences and other repetitive areas, it might also incorporate gene distance 29 Furthermore, the maximum amount of caliber Sanger reads, generally 800-900 bp, in addition to technical problems linked to the sequencing of both DNA stretches with strong secondary structures or extensive homopolymers, make conditions for further sequencing gaps, even in areas with bodily protection.
As many plant genes have conserved areas in their order VIGS may be employed in species, the genomes of that have never been sequenced to some extent. Plant biology poses challenges for the isolation of high quality high-molecular-weight DNA because of strong cell walls, co-purifying polysaccharides, and secondary metabolites that inhibit enzymes or directly damage DNA 14 Consequently, technologies that work nicely on vertebrate genomes might not work well for crops 15 For all these reasons, slow and costly clone-based minimal tiling path sequencing strategies have persisted plants 16, 17 long following quicker, thinner short-read whole-genome assemblies were demonstrated for vertebrate genomes 18 Along with greater genome repetitiveness and dimensions, polyploidy is common in crops (especially crucial crops like cotton, brassicas, wheat, and potatoes) as are high levels of heterozygosity, particularly where inbreeding is debatable as a result of production times 19 or even the plants are obligate outcrossers. The data for chemical systems and genes in plants is stored in the DNA sequences of the genome and the chromosomes.
Colinearity has also been found involving rice and many cereal species, permitting the usage of rice for genetic analysis and gene discovery in more complicated species, including barley and wheat (Shimamoto and Kyozuka, 2002). A comparison out of rice chromosome 3 and regions between barley chromosome 5H showed the presence of four distinct areas, containing four genes. The first deals with the present comprehension of plant genomes, their genetic structure in the inter- and intra- species level and the way entire genomes are sequenced, and its next section addresses some strategies utilised so as to attain the last goal of genomics: discovering the functional and biological significance of DNA sequence. One of the total notes acquired, >99.5percent of the overall reads were plotted on the Bd21 reference genome, and 2.1 million to 3.6 million reads (39.1-50.9% of acquired reads) were plotted on the genomic regions of this 443 SNPs in each sample (Table 2). The accuracy rate of SNP calling by MTA-seq for each accession was between 95.3 and 97.5percent (Table 2), that were in agreement with our whole-genome re-sequencing information of Bd3-1 and Bd21-3, implying that MTA-seq is a viable way of generating amplicons covering p 400 SNP markers in 1 tube, in addition to for its simultaneous genotyping of those amplicons using high-throughput sequencers. DNA sequences of every resulting amplicon were ready from the barley mention genome and united to a single multifasta file for extended bioinformatic analysis (Gupta et al. 2017). GC content for every amplicon was extracted in the multifasta file with the Emboss infoseq instrument (Carver and Bleasby 2003). To tackle these problems and enhance plant genome assemblies, scientists have developed a collection of multifaceted solutions, combining delegated to known public information, like ESTs or BAC ends, or, when available, mention genomes from associated species, integration of genetic and physical map information, or new technology. By way of instance, the meeting of the loblolly pine genome (~22 Gb), that represents the most significant genome constructed thus far, might be solved just with condensed sets and browse pooling before meeting 56 Assembling big and repeat-rich genomes may also be eased by utilizing supplemental layers of data, like the physical space between paired” reads (end-sequences created at either ends of a specific DNA fragment) from mate-pair libraries. An plant genome assembly signifies the entire genomic sequence of these plant species, which can be built into chromosomes and other organelles by utilizing DNA (deoxyribonucleic acid) fragments which are obtained from other kinds of sequencing technology. 4). Target genes were verified by sequencing a randomly chosen PCR product of every plant and hammering the strings at (data not shown). The physical map will be provided by the genomic sequence. To appraise our capacity to detect known variations in selected areas of DNA, we used DNA from 2 non-mutagenized cultivars of linseed flax: CDC Bethune and Macbeth 26 We made primers (Additional file 1) encircling SNVs that was identified in a contrast of CDC Bethune and Macbeth DNA sequences 27 and designated such areas as S20, S411 and S900 with their scaffold of source (e.g. S20 = scaffold 20 of those printed genome assembly two ). We blended DNA from CDC Bethune using DNA from Macbeth to simulate a total of 28 pools from 64 or 96 people, where individual in the swimming pool was polymorphic (i.e. completed a SNV not existing in almost any other member of this pool). The industrial potential of flax, in addition to intriguing facets of its biology (such as well-documented phenotypic and genomic plasticity of a accessions 1), have contributed to a growth in research activity in this species, highlighted by the launch of a meeting of its entire genome sequence 2 to hasten the growth of novel germplasm and to better exploit the available DNA order tools for flax, we sought to develop a mutant people and a reverse osmosis system for this harvest. Since there are genes available to choose from as clones or as sequences for species arrays are created for model organisms like Arabidopsis or rice. Inside this descriptor, we mostly explained the plant substance and complete data sets created and utilized to build, annotate and confirm the tea plant mention genome: (1) raw Illumina entire genome sequencing (WGS) information for genome assembly; (2) raw PacBio sequencing information for genome assembly; (3) raw PacBio RNA sequencing information from mixed cells of tea plant for chemical annotation; (4) eighteen bacterial artificial chromosomes (BACs) and BAC end sequences taken for quality analysis of genome assembly; and (5) that the last assembly and newest release of benchmark genome of tea plant. Generally that the NGS data are utilized in conjunction with Sanger Sequencing technologies or long-reads obtained by the next generation sequencing The genome of this cucumber, (Cucumis sativus), 32 was among those plant genomes that utilizing the NGS Illumina reads in conjunction with Sanger strings. De novo assemblies of plant genomes are performed with NGS reads only, either using scans created over the Illumina platform or with scans created using the Illumina platform along with scans created over the Roche 454 second-generation sequencing stage 45 But, those assemblies are fragmented, leading to low N50 worth and a large number of contigs, largely due to the general brief read span, the complexity of the genome and the existence of conserved areas whose length exceeds the period of NGS reads and consequently cannot be extended throughout the de novo assembly procedure. The assembly has been performed after incorporating extra information derived from cDNA sequences and sequences from subtractive libraries together with methyl-filtered DNA and higher C0t methods, causing a whole-genome assembly (B73 RefGen_v1) manufactured from 2,048 Mb in 125,325 sequence contigs and 61,161 scaffolds 29 Unlike the finished genomes of rice and Arabidopsis, many sequenced BACs from the very first variant of the maize draft genome are incomplete. The complete sequencing of the initial bacterial genomes 15, 16 and the production of initiatives aimed at sequencing the genomes of Sacharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster and Homo sapiens supplied the technological and technical structure for the first sequencing of genomes in plants 17 – 21 These endeavors affirmed the concept of employing a scaled-up kind of shotgun sequencing 22 Shotgun sequencing relied upon computer calculations to empower in silico gathering of overlapping sequencing reads derived from randomly-generated subclones.
Although eliminating primers as far as possible following PCR amplification and before sequencing reactions makes optimum use of sequencing capability by minimizing the research bases utilized for sequencing the primer and optimizing the red bases utilized for sequencing the template, the current inventor discovered that trapping the primers within their entirety contributes to several issues in downstream analysis of sequence information. Sequencing of DNA contained the in depth comparisons of 9 DNA areas utilised to plants that were barcode. The next point was to run NGS-based RAD sequencing to a small number (20) of crops symbolizing that the presence and absence of the gene of interest to yield a high number of sequence reads, followed closely by bioinformatics analysis to discover SNP markers demonstrating correlation between marker genotypes and plant phenotypes.